
Original Article – Haemophilia Volume 14, Issue 6, Pages 1214-1221 – Published Online: 30 Oct 2008 – Reprinted with Permission
N. A. GOLDENBERG*† and M. J. MANCO-JOHNSON* *Hemophilia & Thrombosis Center, Section of Hematology, Oncology, and Bone Marrow Transplantation, Department of Pediatrics, University of Colorado Denver and The Children’s Hospital, Aurora, CO ; and †Division of Hematology/Oncology, Department of Medicine, University of Colorado Denver, Aurora, CO, USA
Correspondence to Marilyn J. Manco-Johnson, Mountain States Regional Hemophilia & Thrombosis Ctr, MS F-416, PO Box 6507, Fitzsimons Bldg 500, Room WG 109, 13001 E. 17th Place, Aurora, CO 80045, USA.
Tel.: 303 724 0365; fax: 303 724 0947; e-mail: marilyn.manco-johnson@uchsc.edu
KEYWORDS
disseminated intravascular coagulation • neonatal thrombosis • protein C • purpura fulminans • thrombophilia
ABSTRACT
Summary. Severe protein C deficiency (i.e. protein C activity <1 IU dL−1) is a rare autosomal recessive disorder that usually presents in the neonatal period with purpura fulminans (PF) and severe disseminated intravascular coagulation (DIC), often with concomitant venous thromboembolism (VTE). Recurrent thrombotic episodes (PF, DIC, or VTE) are common. Homozygotes and compound heterozygotes often possess a similar phenotype of severe protein C deficiency. Mild (i.e. simple heterozygous) protein C deficiency, by contrast, is often asymptomatic but may involve recurrent VTE episodes, most often triggered by clinical risk factors. The coagulopathy in protein C deficiency is caused by impaired inactivation of factors Va and VIIIa by activated protein C after the propagation phase of coagulation activation. Mutational analysis of symptomatic patients shows a wide range of genetic mutations. Management of acute thrombotic events in severe protein C deficiency typically requires replacement with protein C concentrate while maintaining therapeutic anticoagulation; protein C replacement is also used for prevention of these complications around surgery. Long-term management in severe protein C deficiency involves anticoagulation with or without a protein C replacement regimen. Although many patients with severe protein C deficiency are born with evidence of in utero thrombosis and experience multiple further events, intensive treatment and monitoring can enable these individuals to thrive. Further research is needed to better delineate optimal preventive and therapeutic strategies.
Accepted after revision 7 July 2008
DIGITAL OBJECT IDENTIFIER (DOI) 10.1111/j.1365-2516.2008.01838.x About DOI
Introduction
Protein C was isolated from bovine plasma by Johan Stenflo in 1976 and named ‘C’ because it was the third protein to elute from DEAE-Sepharose [1]. However, the function of protein C in the physiological regulation of coagulation was not delineated until several years thereafter. Low levels of plasma protein C were first associated with venous thrombosis in a family study by Griffin et al. in 1982 [2]. The dramatic neonatal presentation of homozygous protein C deficiency with disseminated intravascular coagulation (DIC) and purpura fulminans (PF) within hours of birth was reported by several groups in 1984 [3–5]. These infants and those others subsequently recognized were determined to have a critical defect in coagulation regulation in association with undetectable levels of protein C. Affected infants often died despite frequent infusions of plasma, sometimes because of complications of fluid overload from the amount of plasma required to reverse DIC. Knowledge regarding the molecular and cellular biology of protein C has unfolded over the subsequent years.
Materials and Methods
This article was prepared using published reviews and sentinel source publications. In addition, the authors have personally cared for three persons with severe, moderately severe congenital protein C deficiency from the neonatal period through young adulthood and a large number of children and adults with symptomatic or asymptomatic heterozygous protein C deficiency.
Epidemiology and genetics
The incidence of asymptomatic protein C deficiency has been reported to be between 1 in 200 and 1 in 500 healthy individuals, whereas the incidence of clinically significant protein C deficiency has been estimated at 1 in 20 000 [6]. There is no apparent racial or ethnic predilection for genetic protein C deficiency. Where specific mutations are reported from widely dispersed geographic areas, these reports appear to reflect recurrent mutations related to CG→TG and CG→CA transitions that arise de novo at the highest frequency [7].
Based on a carrier rate of 0.2%, a homozygous or compound heterozygous (i.e. two different allelic mutations) protein C deficiency incidence of 1 per 4 million births could be predicted. However, a recent survey for an FDA pre licensure study of a protein C concentrate (Baxter BioScience, Glendate, CA, USA) identified only 12 living patients with levels of protein C less than 20 IU dL−1 in North America. Potential explanations for the low prevalence of patients with severe genetic protein C deficiency includes excess foetal demise, early postnatal deaths before diagnosis, heterogeneity in the cause of low levels of protein C in the healthy population, and under-reporting.
Cases of individuals with decreased levels of protein C showing familial transmission consistent with heterozygous deficiency have been found among healthy blood donors who had no personal or family history of venous thrombotic events (VTEs) [8,9]. By contrast, two prospective studies of asymptomatic protein C-deficient relatives of protein C-deficient probands showed an increased risk of VTE [10,11]. An investigation based on protein C mutational analysis reported a 50% risk for thrombosis in carriers from symptomatic families by the age of 45 years [12].
The variability in risk of symptomatic VTE in carriers of protein C mutations may be because of incomplete gene penetrance and environmental or genetic cofactors necessary to trigger thrombotic events. Because of the overlap in protein C plasma activity in healthy individuals with those carrying heterozygous protein C gene mutations, it is often difficult to assign carrier status based on a single plasma determination. It has been postulated that a second gene mutation could explain the discrepancy between symptomatic and asymptomatic families with protein C mutations [6,13]. Assayed protein C activity explains some aspects, but not all, of the phenotype. The factor V Leiden mutation accounts for 20% of the variance in white families and investigations are actively exploring other candidate genes.
Most genetic protein C mutations result in type 1 deficiencies in which the decreases in protein C antigen and functional activity are equivalent. Type 2 deficiencies with protein C activity lower than the antigen account for 15% of symptomatic deficiencies [6,13]. To date, there have been more than 160 protein C gene mutations reported [14,15]. There is no single gene mutation that serves as a founder effect causing protein C deficiency in a large number of families. Worldwide, most infants with homozygous protein C deficiency have been born of consanguineous unions and compound heterozygous mutations are more common.
Biology and pathophysiology
Protein C is a vitamin K-dependent coagulation protein that serves a critical role in the regulation of thrombin [see reviews: 6,16–19]. Protein C is synthesized in hepatocytes and circulates in plasma in a very low concentration of approximately 70 nm. Plasma protein C is activated after complex formation with thrombin on the endothelial cell receptor thrombomodulin; this activation is facilitated by protein C binding to the endothelial protein C receptor (EPCR). Activated protein C (APC), augmented by protein cofactors (protein S and factor V) and lipid cofactors (high-density lipoprotein and anionic phospholipids), cleaves critical sites in the activated procoagulant factors V and VIII, thus inactivating these enzymes. Patients with protein C deficiency have a decreased capacity to down-regulate the propagation of thrombin generation by factor Va and VIIIa once they have been activated by the small amounts of thrombin generated in the initiation phase of coagulation activation.
Activated protein C also functions in the regulation of inflammation. During sepsis, signalling by inflammatory cytokines interleukin-1 and tumour necrosis factor mediates altered protein transcription in the systemic inflammatory response (SIR). SIR results in decreased synthesis of the regulatory proteins antithrombin, protein C and protein S, with increased synthesis of prothrombotic proteins factor VIII, von Willebrand factor, and fibrinogen. APC bound to EPCR cleaves the endothelial cell protease activated receptor-1 and, in addition to altered coagulation profiles, causes down-regulation of proinflammatory and proapoptotic mediators, up-regulation of anti-inflammatory and antiapoptotic pathways, and stabilization of endothelial cell barrier functions [16]. The clinical influence of SIR in the pathophysiology of sepsis and the importance of APC in dampening this pathway was demonstrated in the PROWESS trial, in which infusions of recombinant APC resulted in a significant decrease in the mortality of adults with sepsis [20]. Of note, patients with genetic protein C deficiency are not known to have increased susceptibility to sepsis or an altered inflammatory response.
Levels of protein C mature later than many other coagulation proteins. The mean plasma concentration of protein C in a healthy term infant is 40 IU dL−1, with a lower limit of normal of 25 IU dL−1. Protein C concentration increases from birth until 6 months of age when the 50th percentile of paediatric level is equivalent to the 10th percentile of healthy adults (approximately 60 IU dL−1). Protein C concentration remains slightly low through childhood and achieves the adult range after puberty [21]. Healthy adults show a wide observed range of plasma protein C activity of approximately 65–135 IU dL−1 [13]. Authors of this review employ a nomenclature of ‘mild’ protein C deficiency to indicate activity levels greater than 20 IU dL−1 but below the age-appropriate lower limit of normal values, ‘moderately severe’ protein C deficiency as activity levels in the range of 1–20 IU dL−1, and ‘severe’ deficiency for activity levels less than 1 IU dL−1. Most neonatal presentations occur in infants with severe protein C deficiency in whom protein C activity is undetectable. However, rarely, patients with moderate protein C activity have also presented with neonatal PF [22].
Protein C deficiency may be acquired and caused by increased consumption (e.g. overt DIC, severe infection without overt DIC, acute VTE) or by decreased synthesis of the active carboxylated protein (e.g. administration of vitamin K antagonists, severe hepatic synthetic dysfunction, complications of prematurity). Ill preterm infants may have very low levels of protein C activity (e.g. <10 IU dL−1) as an acquired deficiency superimposed on physiologically decreased levels at this age; these low levels may contribute to thrombotic complications in intensively supported preterm infants [21,23]. Rarely, antiphospholipid antibodies (APA) may also cause acquired protein C deficiency via antibody-mediated clearance.
Clinical Manifestations
Infants with severe genetic protein C deficiency usually present within hours of birth with rapidly progressive PF and DIC [24]. PF originates with red or purpuric lesions at pressure points, such as the back of the head and buttocks, as shown in Fig. 1. The lesions rapidly progress to form palpable black eschars that are exquisitely painful. Histologically, PF lesions consist of fibrin clots in small venules of the subcutaneous fat. Coagulation studies are often normal at the outset of skin lesions, except for a markedly elevated D-dimer and an undetectable plasma protein C activity. However, thrombocytopenia, hypofibrinogenaemia, and prolongation of the prothrombin time develop rapidly after onset of PF, if not immediately observed. Other coagulation proteins may be decreased acutely, secondary to consumptive coagulopathy, but normalize to age-appropriate levels after resolution of DIC. Most affected infants manifest white light reflexes and are congenitally blind from thrombosis into the developing vitreal vein and many show evidence of prenatal arterial ischaemic stroke on magnetic resonance imaging of the brain. Large vessel thromboses, including renal vein thrombosis, have been reported in some affected infants. Persons with severe protein C deficiency experience recurrent episodes of PF triggered by infection, trauma, and even minor decreases in goal levels of therapeutic anticoagulation.
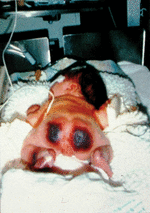
Fig. 1. Compound heterozygous protein C deficiency with undetectable protein C activity. Purpura fulminans in severe protein C deficiency often presents within hours of birth at points of minimal pressure.
A delayed presentation may be observed in adolescents and adults with moderately severe protein C deficiency [25]. The clinical course includes recurrent VTE, including extremity deep vein thrombosis (DVT), pulmonary emboli (PE), parenchymal thrombi and a proclivity to DIC. Although individuals with low levels of detectable protein C often have a clinical presentation that is delayed until puberty, vulnerability of affected individuals to DIC and thrombosis thereafter may be similar to that seen in patients with neonatal presentation.
The clinical phenotype of simple heterozygous protein C deficiency, characterized by mild deficiency in measured protein C activity, can range from asymptomatic to a potent thrombophilic state with recurrent thromboses resulting in severe venous insufficiency from the post-thrombotic syndrome, as shown in Fig. 2. In addition to DVT and PE, the patients with heterozygous protein C deficiency may develop ischaemic arterial stroke, mesenteric thrombi and pregnancy-associated thrombosis [26–28]. Patients with a significant positive family history, multiple thrombophilia traits, APA, or underlying inflammatory disorders are more likely to develop thrombotic manifestations, while more benign personal and family histories are often characterized by mild protein C deficiency as a single thrombophilic defect.
Fig. 2. Symptomatic heterozygous protein C deficiency. Individuals with recurrent venous thromboembolism secondary to heterozygous protein C deficiency are at risk for post-thrombotic syndrome. This patient manifests venous stasis ulcers.
Diagnosis
Diagnostic testing for protein C deficiency typically uses functional assays. Chromogenic assays for protein C that use activation by snake venom in an activating reagent (Protac; Aniara Corp, Mason, OH, USA) are widely available. Clotting assays and enzyme-linked immunosorbent assays are also commercially available. Given the occurrence of acquired deficiency, retesting of patients with low protein C levels to exclude a transient deficiency is recommended after resolution of acute consumptive states and when not receiving oral anticoagulant therapy. Patients with APA and low levels of protein C can be tested for antiprotein C antibodies to determine whether protein C deficiency is congenital or acquired. Quality assurance issues in sample collection, assay performance, and interpretation (i.e. preanalytic and analytic conditions) are extremely important in the determination of protein C activity [29].
In the case of suspected homozygous or compound heterozygous protein C deficiency, testing both parents and all grandparents can be helpful. Most parents of infants with severe protein C deficiency are asymptomatic and have protein C levels compatible with heterozygous status.
Protein C mutational analysis is available in a few laboratories; see persons interested in protein C sequencing below. Results of DNA studies can be useful for confirmation of carrier status or for prenatal diagnosis. Prenatal diagnosis can be made by mutational analysis of the protein C gene using chorionic villous sampling material or amniotic cells. For the protein C mutation database, readers are referred to the Web site of the Scientific Standardization Committee of the International Society for Thrombosis and Haemostasis, Subcommittee on Plasma Coagulation Inhibitors, currently chaired by Dr. Elaine Gray (http://www.med.unc.edu/isth/ssc_home.htm).
Management
Neonatal PF can be controlled only with protein C replacement in the form of fresh frozen plasma (FFP) or a human plasma–derived, viral inactivated protein C concentrate [30–32]. A human plasma-derived, viral-inactivated protein C concentrate manufactured by Baxter (Ceprotin®; Baxter BioScience, Glendale, CA, USA) has been licensed in the United States and Europe. A second plasma-derived concentrate (Protexel®; LFB, Lille, France) is also available in Europe. Protein C replacement dosing is similar to that for other vitamin K-dependent proteins, such as factor IX. A total of 1 IU kg−1 of protein C concentrate or 1 mL kg−1 of FFP will increase the plasma concentration by 1 U dL−1. Based on a 6- to 10-h half-life of protein C in plasma in the steady state, patients with severe protein C deficiency who have DIC or acute thrombosis can be treated acutely with an initial bolus of 100 IU kg−1 followed by 50 IU kg−1 every 6 h. Author recommendations for management of patients with severe protein C deficiency are given in Table 1.
Table 1. Author recommendations for management of patients with severe genetic protein C deficiency.
| Condition | Treatment product | Dosing | Target therapeutic indicator |
|---|---|---|---|
|
|
|||
| DIC, PF, acute VTE | Protein C concentrate | 100 U kg−1 initial bolus; 50 U kg−1 every 6–12 h (every 6 h acutely) | Protein C activity 50 IU dL−1 trough; decreasing (or normalization of) D-dimer |
| Fresh frozen plasma | 10–15 mL kg−1, every 8–12 h, until protein C concentrate available | Protein C activity >10 IU dL−1 trough, until protein C concentrate available; decreasing (or normalization of) D-dimer | |
| Unfractionated heparin* | 15–20 U kg−1 h−1 with protein C replacement | Anti-Xa activity 0.3–0.7 U mL−1 | |
| Low-molecular-weight heparin* | 1.0–1.5 mg kg−1 every 12 h with protein C replacement | Anti-Xa activity 0.5–1.0 U mL−1 | |
| Prophylaxis for invasive procedures | Protein C concentrate | 100 U kg−1 initial bolus; 30–50 U kg−1 every 12–24 h | Protein C activity 20–50 IU dL−1 trough; negative (or rapidly decreasing) D-dimer in postoperative period |
| Maintenance | Protein C concentrate (typically with oral anticoagulation, below) | 30–50 IU kg−1 every 1–3 days | D-dimer negative |
| Warfarin | 0.1–0.2 mg kg−1 orally every day (without protein C replacement); 0.05–0.1 mg kg−1 orally every day with protein C replacement | INR 2.5–3.5, higher INR typically required in adolescents, INR 1.52.5 with protein C replacement | |
| DIC, disseminated intravascular coagulation; INR, international normalized ratio; PF, purpura fulminans; VTE, venous thromboembolism. | |||
| *May pose an increased bleeding risk in the setting of thrombocytopenia in DIC. | |||
Outside of DIC, PF, or an acute VTE event, most affected infants have been managed for long-term secondary prophylaxis with protein C concentrate or therapeutic anticoagulation using either low-molecular-weight heparin or high-intensity warfarin [30–34]. Warfarinization should always be preceded by several days of therapeutic heparin administration to avoid warfarin skin necrosis and other progressive or recurrent thrombotic complications, including VTE, PF, or DIC. Alternatively, children can be managed with a combination of the two approaches (e.g. protein C replacement, approximately 50 U kg−1, given every other day or three times weekly in combination with less intensive anticoagulation). Monitoring for evidence of coagulation activation with D-dimer is useful to confirm adequate replacement or anticoagulation therapy; in the authors’ experience, and as previously published in the neonate [33], a markedly elevated or rapidly rising D-dimer has often heralded the onset of recurrent VTE, PF, or DIC in young patients with severe protein C deficiency.
Recombinant APC (Xigris; Eli Lilly, Indianapolis, IN, USA) has been used by the authors to treat PF in a child with severe protein C deficiency. Recombinant APC, administered at a dose of 24 μg kg−1 h−1 (the regimen recommended for sepsis), resulted in a plasma protein C activity of 5 IU dL−1 and has been variably effective in controlling PF [35]. However, a non-significant trend to increase in major bleeding episodes in a paediatric randomized controlled trial of recombinant APC in sepsis (particularly among young infants) [36] has raised concern regarding the potential haemorrhagic risk of this agent in children.
Complications of therapy
The use of FFP can be complicated by fluid overload, as noted above. In addition, FFP is not available in the USA as a viral-inactivated product. Furthermore, exposure to large numbers of donors over time increases the cumulative risk for transfusion-associated viral infection and allergic reaction to donor proteins in FFP. By contrast, protein C concentrate has a small theoretical risk of viral contamination, similar to other Food and Drug Administration-approved human plasma-derived, viral inactivated protein concentrates. Allergic reactions and alloantibody formation are potential complications of any protein replacement therapy but have not been described in patients to date.
Long-term replacement therapy in small infants has required placement of central venous access devices. Because of the inherent hypercoagulability of affected patients, thrombotic and infectious complications of central venous access devices present limitations to continuous replacement therapy and must be accompanied by strict adherence to the protein C replacement regimen, in addition to any concomitant low-dose anticoagulation. Routine instillation of a fibrinolytic agent into the central venous access device may be helpful to prevent catheter occlusions and infections, but there is no evidence to support this practice.
Prognosis
Published data on the long-term outcome of persons affected with severe genetic protein C deficiency are limited. For this reason, at present, information on outcomes is principally limited to anecdotal experiences. Point-of-care testing for international normalized ratio with home monitors has greatly facilitated care of patients with severe genetic protein C deficiency. Early recognition of subtherapeutic international normalized ratios (INR), accompanied by institution of short-term bridging anticoagulation with low-molecular-weight heparin or protein C replacement with concentrate, can successfully prevent recurrent episodes of VTE, DIC and PF. With proactive home monitoring and patient-directed management, the need for hospitalization can become infrequent. For example, a 20-year-old woman managed by the authors with severe protein C deficiency and congenital blindness because of bilateral vitreal vein thrombosis in utero, who adeptly uses a home INR monitoring device, has been hospitalized three times in the past six years including twice for bleeding episodes on high-dose warfarin (target INR: 3.5–3.8), and once for Mediport placement to permit regular protein C replacement as a means of reducing chronic anticoagulant dose and, hence, the risk of future bleeds. This patient has been free of hospitalization for the past 2 years while on a prophylactic thrice-weekly protein C replacement regimen in conjunction with low-dose warfarin.
The cumulative likelihood of severe bleeding complications increases with the duration of long-term anticoagulation. Of considerable concern is the risk of ruptured ovarian cyst with resultant significant pelvic haemorrhage among adolescent and young adult females receiving long-term anticoagulation, and traditional suppressive therapy with oestrogens is contraindicated in patients with protein C deficiency, unless protein C is being adequately replaced. In addition, surgical intervention for the management of viscous thromboembolic complications, such as bowel infarct, also poses a challenge with regard to the simultaneous risks of severe haemorrhage and recurrent thrombotic events. The availability of protein C concentrate has been judged to be life-saving in such circumstances.
Each of three children with severe/moderately severe genetic protein C deficiency managed and followed long-term by the authors has been adequately supported through major surgery with protein C replacement. No anticoagulation has been required when plasma protein C concentrations have been maintained above a nadir of 50% around surgery and above 20% during baseline conditions; this has enabled invasive surgical procedures to be performed with low risk of major haemorrhage.
Some patients with simple heterozygous protein C deficiency, including one followed by the authors, have experienced recurrent severe episodes of DVT, leading to advanced post-thrombotic syndrome with venous stasis ulcers that heal poorly and are difficult to manage. The coexistence of obesity is an important candidate prognostic factor for the development of post-thrombotic syndrome in patients with thrombophilia.
The lupus anticoagulant has developed in patients with protein C deficiency and recurrent thrombosis, including two of the three severe/moderately severe protein C-deficient patients managed by the authors. Although the relationship of thrombophilia to development of APA is unknown, it is possible that vascular damage contributes to the development of the antibodies. Our experience suggests that APA may exacerbate the prothrombotic state and clinical course in protein C-deficient individuals.
It is worthy of note that osteoporosis has developed in the 20-year-old woman described earlier, maintained on life-long intensive oral anticoagulation with warfarin. Given that oral anticoagulation with vitamin K antagonists is not associated with osteoporosis, it is likely that long-term decreases in normal impact physical activities because of congenital blindness contributed to decreased bone density in this patient. Heightened surveillance of bone mineral density in congenitally blind protein C-deficient patients and early institution of isometric exercise regimens and medication for osteoporosis may be warranted to prevent fractures in such patients, who are at risk for both thrombotic and haemorrhagic complications with orthopaedic injury.
With regard to physical and cognitive development, all three children with severe/moderately severe protein C deficiency managed by the authors with intensive anticoagulation and/or protein C replacement have exhibited normal growth in long-term follow-up. Furthermore, although some reported children with severe protein C deficiency have manifest developmental delays and/or cognitive impairment, these three patients have achieved above average academic success in university and graduate school programmes.
Individuals with interest in the area
Paediatric haematology
Marilyn J. Manco-Johnson, MD; email: marilyn.manco-johnson@uchsc.edu; Neil A. Goldenberg, MD, PhD; email: neil.goldenberg@uchsc.edu; Tel. (both) 303 724 0365.
Protein C gene sequencing
Craig Hooper, PhD, United States Centers for Disease Control and Prevention, 1600 Clinton Rd., Bldg 1 Room 1332, Atlanta, GA 30333, USA; Tel.: 404 639 3750; Fax: 404 639 1638; email: woh1@cdc.gov. Dr Hooper could provide mutation analysis as part of a research/registry project. The time to results is dependent on the number of samples submitted for analysis.
The laboratory of Dr Christine Mannhalter is licensed for genetic testing and can provide mutation analysis for protein C deficiency. The time to results ranges between two and three weeks per sample. Contact information for Dr Mannhalter is as follows: Univ.-Prof. Dr Christine Mannhalter, Klinisches Institut für Medizinische & Chemische Labordiagnostik, Medizinische Universität Wien, Allgemeines Krankenhaus, Wien. Währinger Gürtel 18-20, A-1090 Wien, Tel.: (+43/1) 40 400 20 85, Fax: (+43/1) 40 400 20 97, e-mail: christine.mannhalter@meduniwien.ac.at.
Links to organizations that can provide information, referrals and support
National Blood Clot Alliance (NBCA) is an advocacy society for persons and families dealing with thrombosis and thrombophilia:http://www.stoptheclot.org.
The Anticoagulation Forum is a network of healthcare professionals committed to the therapy of thromboembolic disorders predominantly through the venue of anticoagulation management service: http://www.acforum.org.
The United States Centers for Disease Control and Prevention Pilot Program in Thrombophilia and Thrombosis:http://www.cdc.gov/ncbddd/hbd/thromb_center_list.htm and http://www.cdc.gov/ncbddd/hbd/clotting.htm.
Conclusions and future directions
Severe protein C deficiency remains a rarely diagnosed condition. It is possible that affected individuals may experience excess foetal demise or that affected infants die from DIC before assignment of the diagnosis. Given the availability of safe, effective replacement proteins, renewed educational outreaches to paediatricians, obstetricians, and primary care physicians may help to identify affected infants who would benefit from such therapies. At the same time, future study of dosing, efficacy, and safety of protein C replacement for both prophylaxis and therapy in identified individuals with severe and moderately severe protein C deficiencies is urgently needed. Given the rarity of these protein C deficiency states, and yet their frequently severe impact on longevity and quality of life, strong advocacy efforts will likely be necessary to continue active research efforts (both observational and interventional) directed toward an ultimate goal of optimizing care of affected patients.
Disclosures
The authors stated that they had no interests which might be perceived as posing a conflict or bias
References
- Stenflo J. A new vitamin K-dependent protein: purification from bovine plasma and preliminary characterization. J Biol Chem 1976; 251: 355–63. Links
- Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. J Clin Invest 1981; 68: 1370–3. Links
- Seligsohn U, Berger A, Abend M et al. Homozygous protein C deficiencty manifested by massive venous thrombosis in the newborn. N Engl J Med 1984; 310: 559–62. Links
- Estelles A, Garcia-Plaza I, Dasi A et al. Severe inherited “homozygous” protein C deficiency in a newborn infant. Thromb Haemost 1984; 52: 53–6. Links
- Sills RH, Marlar RA, Montgomery RR, Deshpande GN, Humbert JR. Severe homozygous protein C deficiency. J Pediatr 1984; 105: 409–13. Links
- Dahlbäck B. The protein C anticoagulant system: inherited defects as basis for venous thrombosis. Thromb Res 1995; 77: 1–43. Links
- Gomez K, Laffan MA. Hunting for the mutation in inherited thrombophilia. Blood Coagul Fibrinolysis 2004; 15: 125–7. Links
- Tait RC, Walker ID, Reitsma PH et al. Prevalence of protein C deficiency in the healthy population. Thromb Haemost 1995; 73: 87–93. Links
- Miletich J, Sherman L, Broze G Jr. Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med 1987; 317: 991–6. Links
- Henkens CM, van der Meer J, Hellege JL, van der Schaaf W, Bom VJ, Halie MR. The clinical expression of hereditary protein C and protein S deficiency: a relation to clinical thrombotic risk factors and to levels of protein C and protein S? Blood Coagul Fibrinolysis 1993; 4: 555–62. Links
- Pabinger I, Kyrle PA, Heistinger M, Eichinger S, Willmann E, Lechner K. The risk of thromboembolism in asymptomatic patients with protein C and protein S deficiency: a prospective cohort study. Thromb Haemost 1994; 71: 441–5. Links
- Allaart CR, Poort SR, Rosendaal FR, Reitsma PH, Bertina RM, Briët E. Increased risk of venous thrombosis in carriers of hereditary protein C deficiency defect. Lancet 1993; 341: 134–8. Links
- Bertina RM. Protein C deficiency and venous thrombosis: the search for the second genetic defect. Thromb Haemost 2000; 83: 360–1. Links
- Reitsma PH, Bernardi F, Doig RG et al. Protein C deficiency: a database of mutations, 1995 update. On behalf of Subcommittee on plasma coagulation inhibitors of the SSC of the ISTH. Thromb Haemost 1995; 73: 876–9. Links
- Alhenc-Gelas M, Gandrille S, Aubry M-L, Aiach M. Thirty-three novel mutations in the protein C gene. Thromb Haemost 2000; 83: 86–92. Links
- Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood 2007; 109: 3161–72. Links
- Dalbäck B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway : novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol 2005; 25: 1311–20. Links
- Esmon CT. The protein C pathway. Chest 2003; 124: 26–32. Links
- Esmon C. The protein C pathway. Crit Care Med 2000; 28: S44–8. Links
- Cobas MM, Langenfeld H, Rossaint R, Sablotzki A. PROWESS, ENHANCE, and ADDRESS: clinical implications for the treatment with drotrecogin alfa (activated). Anaesthetist 2006; 55(Suppl. 1): 16–23. Links
- Petäjä J, Manco-Johnson MJ. Protein C pathway in infants and children. Semin Thromb Hemost 2003; 29: 349–61. Links
- Tcheng WY, Dovat S, Gurel Z, Donkin J, Wong W-Y. Severe congenital protein c deficiency: description of a new mutation and prophylactic protein C therapy and in vivo pharmacokinetics. J Pediatr Hematol Oncol 2008; 30: 166–71. Links
- Manco-Johnson MJ, Abshire TC, Jacobson LJ, Marlar RA. Severe neonatal protein C deficiency: prevalence and thrombotic risk. J Pediatr 1991; 119: 793–8. Links
- Marlar RA, Montgomery RR, Broekmans AW. Diagnosis and treatment of homozygous protein C deficiency. Report of the working party on homozygous protein C deficiency of the Subcommittee on Protein C and Protein S, International Committee on Thrombosis and Haemostasis. J Pediatr 1989; 114: 528–34. Links
- Zimbelman J, Lefkowitz J, Schaeffer C et al. Unusual complication of warfarin therapy: skin necrosis and priapism. J Pediatr 2000; 137: 266–8. Links
- Kohler J, Kasper J, Witt I, von Reutern GM. Ischemic stroke due to protein C deficiency. Stroke 1990; 21: 1077–80. Links
- Yates P, Cumber PM, Sanderson S, Harrison BJ. Mesenteric venous thrombosis due to protein C deficiency. Clin Lab Haematol 1991; 13: 137–9. Links
- Pabinger I, Grafenhofer H. Thrombosis during pregnancy: risk factors, diagnosis and treatment. Pathophysiol Haemost Thromb 2002; 32: 322–4. Links
- Mackie I, Cooper P, Kitchen S. Quality assurance issues and interpretation of assays. Semin Hematol 2007; 44: 114–25. Links
- Dreyfus M, Masterson M, David M et al. Replacement therapy with a monoclonal antibody purified protein C concentrate in newborns with severe congenital protein C deficiency. Semin Thromb Hemost 1995; 21: 371–81. Links
- [No authors listed.] Human protein C: new preparations: effective replacement therapy for some clotting disorders. Prescrire Int 2003; 12: 11–3. Links
- Hartman KR, Manco-Johnson M, Rawlings JS, Bower DJ, Marlar RA. Homozygous protein C deficiency: early treatment with warfarin. Am J Pediatr Hematol Oncol 1989; 11: 395–401. Links
- Monagle P, Andrew M, Halton J et al. Homozygous protein C deficiency: description of a new mutation and successful treatment with low molecular weight heparin. Thromb Haemost 1998; 79: 756–61. Links
- Dreyfus M, Magny JF, Bridey F et al. Treatment of homozygous protein C deficiency and neonatal purpura fulminans with a purified protein C concentrate. N Engl J Med 1991; 325: 1565–8. Links
- Manco-Johnson MJ, Knapp-Clevenger R. Activated protein C concentrate reverses purpura fulminans in severe genetic protein C deficiency. J Pediatr Hematol Oncol 2004; 26: 25–7. Links
- Nadel S, Goldstein B, Williams MD et al. Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet 2007; 369: 836–43. Links
Posted November 15, 2008



